
Research projects
Simulating atomistic model of OMEICs under constant electrochemical potential method
We developed an innovative simulation protocol of QM/MM + Grand Canonical scheme + classical molecular dynamics simulation (QM/MM + GCMD) where redox species in solution are in equilibrium with an electrode at a controllable electrochemical potential (Φ). Based on this protocol, we study how the microstructure and charge distribution change in the polymer in OMEICs under different Φ.
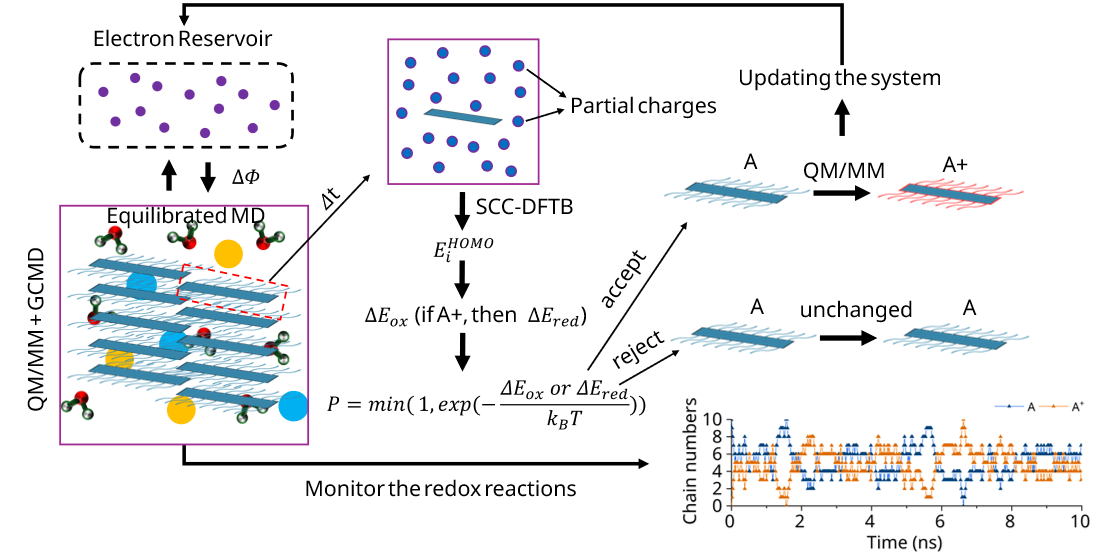
Highlighted related publications:
- Wei, Z*., Makki, H., Troisi, A*. (2025). The relation between the doping level and structures in semicrystalline polymeric ionic-electronic conductors under the constant electrochemical potential. In preparation.
- Wei, Z*., Makki, H., Carbone, P*., Troisi, A*. (2025). Simulation of charge distribution and microstructure in semicrystalline polymeric ionic-electronic conductors using classical simulation and constant electrochemical potential. Manuscript.
Simulating atomistic model of supercapacitors under constant surface charges method
By combining innovative QM/MD simulations with experimental data, we explained how electrowetting and total capacitance are driven by interfacial hydrogen bond networks and electric double layer structures, respectively.
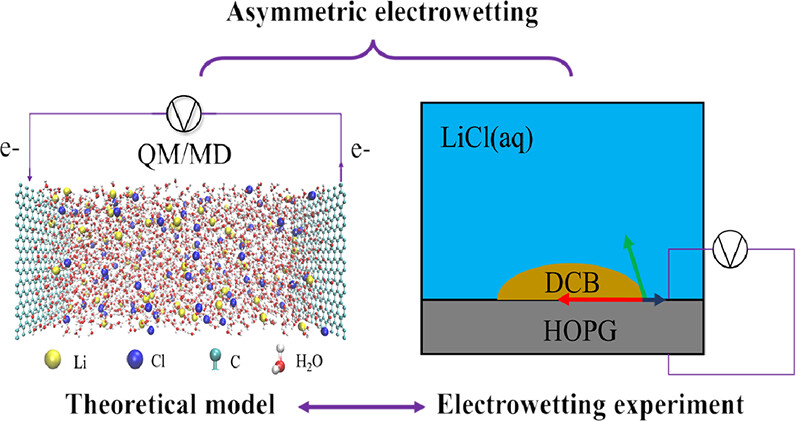
Highlighted related publications:
- Wei, Z., Elliott, J., Papaderakis, A*., Dryfe, R*., Carbone, P*. Relation between double layer structure, capacitance and surface tension in electrowetting of graphene and aqueous electrolytes. J. Am. Chem. Soc 146, 760-772, (2024). DOI: 10.1021/jacs.3c10814
- Papaderakis, A*,1., Leketas, M1., Wei, Z1., Hwang, I1., Carbone, P1., Juel, A1., Dryfe., R1. Electrowetting of carbon-based materials for advanced electrochemical technologies. ChemElectroChem 11, e202400143, (2024). DOI: 10.1002/celc.202400143 (co-first author)
Investigating water behavior under 2D confinement
We revealed water behavior (structure and dynamics) at the interface and the stability of 2D material nanochannels under different levels of cofinement via MD simulation.
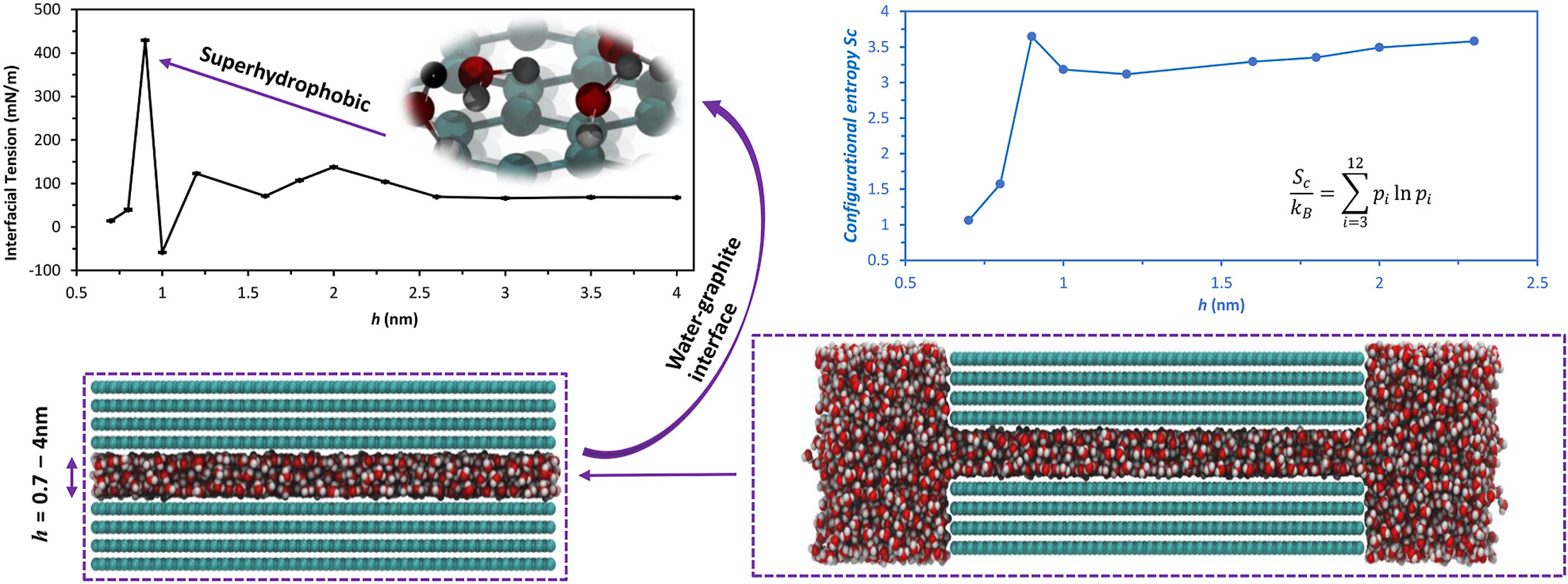
Highlighted related publications:
- Smith, L., Wei, Z., Williams, C., Chiricotto, M., Fonte, C., Carbone, P*. Relationship between Capillary Wettability, Mass and Momentum Transfer in Nano-Confined Water: the Case of Water in Nanoslits of Graphite and Hexagonal Boron Nitride. ACS Applied Materials & Interfaces, (2024). DOI: 10.1021/acsami.4c10738
- Wei, Z., Chiricotto, M., Elliott, J., Martelli, F., Carbone, P*. Wettability of graphite under 2D confinement. Carbon 198, 132-141, (2022). DOI: 10.1016/j.carbon.2022.07.019
- Williams, C. D., Wei, Z., Shaharudin, M. R. bin & Carbone, P*. A molecular simulation study into the stability of hydrated graphene nanochannels used in nanofluidics devices. Nanoscale 14, 3467–3479 (2022). DOI: 10.1039/d1nr08275b
Simulating SEI formation in the Solid-state battery (a future plan)
Simulating Solid–Electrolyte Interphase (SEI) formation remains a challenging problem in computational chemistry. I aim to combine Monte Carlo and machine-learning approaches with molecular dynamics (MD) simulations to model SEI formation under a constant electrochemical potential in solid-state batteries.